
Epithelial-Mesenchymal Transition Contributes to Pulmonary Fibrosis via Aberrant Epithelial/Fibroblastic Cross-Talk
Charlotte Hill1, Mark G. Jones2,3, Donna E. Davies2,3,4 and Yihua Wang1,4*
1Biological Sciences, Faculty of Environmental and Life Sciences, University of Southampton, Southampton SO17 1BJ, UK.
2Clinical and Experimental Sciences, Faculty of Medicine, University of Southampton, Southampton SO16 6YD, UK.
3NIHR Respiratory Biomedical Research Centre, University Hospital Southampton, Southampton SO16 6YD, UK.
4Institute for Life Sciences, University of Southampton, Southampton SO17 1BJ, UK.
Abstract
Idiopathic pulmonary fibrosis (IPF) is the prototypic progressive fibrotic interstitial lung disease. Median survival is only 3 years, and treatment options are limited. IPF is thought to be a result of a combination of genetic and environmental factors with repetitive micro-injuries to alveolar epithelial cells playing a central role. IPF is characterised by aberrant extra cellular matrix (ECM) deposition by activated myofibroblasts. Epithelial-mesenchymal transition (EMT) is a process where polarised epithelial cells undergo molecular changes allowing them to gain a mesenchymal phenotype, with a subsequent enhanced ability to produce ECM components and increased migration and/or invasion. The source of myofibroblasts in IPF has been debated for many years, and EMT has been proposed as a source of these cells. However, lineage tracing in transgenic mice suggests the contribution of epithelial cells, which have undergone EMT, to the fibroblast population may be negligible. Instead, recent findings suggest that alveolar epithelial type II (ATII) cells undergoing EMT promote a pro-fibrotic microenvironment through paracrine signalling activating local fibroblasts. This review paper explores the contribution of ATII cells, which have undergone EMT, in the context of pulmonary fibrosis.
Introduction and Discussion
Idiopathic pulmonary fibrosis (IPF)
IPF is a chronic, progressive and fibrotic lung disease of unknown cause, which typically occurs in older adults. Alveolar architecture is destroyed and healthy tissue is replaced by altered extra cellular matrix (ECM), with progressive dyspnoea and impairment of lung function ultimately leading to death1,2. IPF is the most common type of idiopathic interstitial pneumonia and occurs with similar frequency to stomach, brain and testicular cancer3. Although the cause of IPF is unknown, interacting genetic and environmental factors are thought to play a role in its development2. Repetitive injury to aged alveolar epithelium is proposed to trigger aberrant wound healing processes, initiating an accumulation of ECM deposited by myofibroblasts2. Current approved therapies only slow IPF disease progression highlighting the need for better understanding of the disease process and the identification of new molecular targets.
Epithelial-mesenchymal transition (EMT)
EMT is a dynamic, reversible process which has been implicated in embryonic development, wound healing, cancer metastasis and fibrosis and is associated with an increased migratory and/or invasive ability4. The role of EMT in cancer is detrimental whereas, in wound healing, EMT as a response to injury can be beneficial, however, if the wound healing process is exaggerated it may lead to fibrosis. During EMT, epithelial cells lose apical-basal polarity, tight and adherens junctions in favour of front-back polarity, N-cadherin junctions and vimentin stress fibres4,5. The change in morphology is accompanied by molecular change initiated by several pathways and signalling factors which regulate expression of transcription factors (EMT-TFs), including Snail, ZEB, Twist and others6. Pleotropic signalling factors such as transforming growth factor β (TGFβ), fibroblast growth factor (FGF), Wnt/β-catenin and epidermal growth factor (EGF) can initiate EMT, in turn, these factors regulate expression of EMT-TFs. These promote the repression of epithelial features by suppressing E-cadherin expression, and induction of mesenchymal features, in part, through the activation of mesenchymal genes N-cadherin, vimentin and fibronectin, which are responsible for cell-cell adhesion, cell motility, and migration6–11.
Induction of EMT in fibrosis has been linked to a variety of processes including endoplasmic reticulum (ER) stress, smoking, Epstein Barr virus protein LMP1 (latent membrane protein 1)12–14 and EGF receptor (EGFR) signalling15. A number of studies have implicated EGFR activation16–18, mutations19 and increased expression20 in IPF patients. An increase in transforming growth factor α (TGFα) and EGFR in rats with bleomycin-induced lung injury16 has been observed. TGFα was also increased in response to asbestos and hyperoxia17,18. Transgenic mice that constitutively express TGFα develop progressive and severe lung fibrosis21, chronic expression of TGFα in new born transgenic mice resulted in remodelling of lung during postnatal alveolarisation resulting in pulmonary fibrosis22. Further, mice treated with inhibitors of the EGFR pathway display resistance to bleomycin-induced fibrosis23. Gene network analysis of publicly available microarray data (GSE24206) of IPF and control lung tissue has identified the EGFR-ERK pathway to be a top-ranked pathway15. Further, RAS signalling is a key pathway downstream of EGFR activation and RAS activation has been demonstrated to induce EMT in other contexts24,25. Together, these results highlight the potential importance of EGFR activation in IPF. Elucidating the downstream mechanisms and processes which could be activated as a result of this, may provide new targets for treatment. Of relevance to IPF is the demonstration that EGFR activation induces EMT in ATII cells, where they undergo a change in morphology, with reorganisation of actin cytoskeleton, accompanied by an increase in vimentin, ZEB1 and a reduction in E-cadherin15. It has been demonstrated through inhibition of AKT or ERK that RAS activation induces EMT in ATII cells via ERK pathway. Further, RNA interference (RNAi)-mediated knockdown of EMT-TFs confirmed that RAS-induced EMT in ATII cells is specifically via ZEB115. Consistent with this, analysis of human IPF lung tissue demonstrates that in comparison with control lung tissue, strong nuclear staining of ZEB1 is present in fibroblast foci15,26,27 and also in epithelial cells of thickened alveoli septae, where collagen deposition in the interstitium is evident15.
Contribution of EMT to pulmonary fibrosis
In the distal region of the lung there are two types of alveolar epithelial cells; type I and type II, with the first providing thin-walled gas-exchange surface and the latter functions as stem cells, contributing to alveolar renewal and repair28. The origin of myofibroblasts in IPF is controversial but it has been proposed that ATII cells that have undergone EMT may be a source of myofibroblasts in fibrosis. Myofibroblasts are understood to be critical in the pathogenesis of IPF, with increased fibroblast foci associated with worse prognosis29.
Human IPF tissue demonstrated co-localisation of epithelial and mesenchymal markers26,27,30–32. Laser capture microdissection has also been performed to isolate RNA from epithelial cells in IPF lungs which confirmed expression of mesenchymal markers by epithelial populations33, suggesting that EMT may contribute to the mesenchymal population. However, in mouse models of lung fibrosis, the conclusions of lineage tracing studies investigating the contribution of EMT to the mesenchymal population have been varied. Several studies have suggested that EMT may contribute to the pathogenesis of IPF in vivo34-39. Conversely, other lineage tracing studies found relatively small numbers of fibroblasts arise from epithelial cells40,41. In addition, it was shown that α-smooth muscle actin (α-SMA) did not co-localise with EMT-derived cells, suggesting that although they may have undergone EMT, they did not transition to myofibroblasts36,37. Similarly, we reported that ATII cells which have undergone RAS-induced EMT produce extremely low levels of ECM genes15. Thus, the significance of EMT and these cells’ ability to contribute towards the mesenchymal population are still somewhat controversial.
Studies in renal fibrosis have proposed that tubular epithelial cells are able to promote myofibroblast differentiation and fibrogenesis without directly contributing to the population by relaying signals to the interstitium. It was demonstrated that reactivation of Snail1 in renal epithelial cells was required for the development of fibrosis in the kidney. In a murine model, damage-mediated Snail1 reactivation induced EMT but these cells did not contribute to myofibroblast or interstitial cell population. Epithelial cells which have undergone EMT did subsequently relay signals to the interstitium to promote myofibroblast differentiation and fibrogenesis42. In mouse models of experimentally induced renal fibrosis, Snai1 or Twist1 deletion led to inhibition of EMT, while restoring proliferation, repair and regeneration ultimately attenuated interstitial fibrosis43. In renal fibrosis TGFβ induces EMT via Snail1 (SNAI1), in turn, this induces TGFβ expression generating an autocrine loop sustaining myofibroblast differentiation42. Snail1 has also been demonstrated to be up-regulated in mouse models of acute liver fibrosis during tissue remodelling44.
In the context of pulmonary fibrosis, it has also been reported that ATII cells appear to promote fibrogenesis without direct contribution to the population. We demonstrated that conditioned media (CM) from RAS-activated ATII cells was able to potentiate fibroblast activation in the presence of TGFβ, which could be produced as a result of damage to alveolar epithelial cells. RNAi-mediated knockdown of ZEB1 abolished the effects of the RAS-activated CM on fibroblast, therefore ZEB1 was demonstrated to be a key regulator of the paracrine signalling between alveolar cells and fibroblasts15. Further, it was determined that ZEB1 controls tissue plasminogen activator (tPA) expression, which subsequently affects fibroblast activation induced by TGFβ15. tPA has previously been identified in the context of kidney fibrosis and was shown to promote TGFβ-mediated α-SMA and type I collagen expression45. Quantitative analysis of CM from RAS-activated ATII cells identified secreted proteins, expression of these was then compared with a publicly available dataset46 and 25 genes/proteins were identified in both. PLAT which encodes tPA was the most up-regulated in ATII cells, and was identified in this list. A ZEB1 binding site was identified in the promoter region of PLAT, mRNA expression of PLAT was increased upon RAS-activation and this was repressed by ZEB1 RNAi15.
In both pulmonary and kidney fibrosis, it appears that although epithelial cells do not directly contribute to myofibroblast populations via EMT, they are able to promote myofibroblast differentiation through secreted factors (Fig. 1), and that these could potentially be the source of novel targets of treatment.
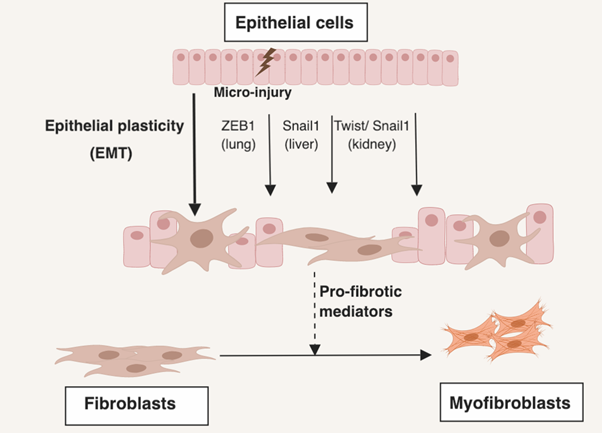
Figure 1: EMT of epithelial cells dysregulates signalling between epithelial and mesenchymal cells leading to a pro-fibrotic microenvironment. Repetitive micro-injury to epithelial cells creates a pro-fibrotic environment. A number of EMT transcription factors (EMT-TFs) have been demonstrated to induce EMT programs in a variety of tissues in the context of fibrosis. In pulmonary fibrosis, ZEB1 is responsible for RAS-induced EMT15. In the liver Snail1 has been demonstrated to induce EMT44. In the context of renal fibrosis, Snail1 and Twist1 have been demonstrated to induce EMT42,43. However, lineage tracing studies suggest these fibroblasts do not contribute significantly to the myofibroblast population40,41. These EMT-TFs do appear to mediate creating a pro-fibrotic microenvironment15,42,43.
Conclusion
Epithelial cells undergoing EMT produce relatively low levels of ECM and lineage-tracing studies have demonstrated that they do not significantly contribute directly towards the mesenchymal population. Recent studies have identified that across organs, EMT may instead promote a pro-fibrotic microenvironment by dysregulating paracrine signalling between epithelial and mesenchymal cells. Targeting EMT inducers might have therapeutic potential in fibrotic conditions, with such therapies currently undergoing development in the context of malignancy47,48.
Acknowledgements
This project was supported by the Academy of Medical Sciences/the Wellcome Trust Springboard Award [SBF002\1038], Wessex Medical Trust and AAIR Charity. CH was supported by Gerald Kerkut Charitable Trust and University of Southampton Central VC Scholarship Scheme. MGJ was supported by the Wellcome Trust [100638/Z/12/Z].
References
- Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2018; 198: e44–e68.
- Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017; 389: 1941–1952.
- Hutchinson J, Fogarty A, Hubbard R, et al. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J. 2015; 46: 795–806.
- Nieto, A., Huang, R., Jackson, R. et al. EMT: 2016. Cell. 2016; 166: 21–45.
- Onder TT, Gupta PB, Mani SA, et al. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008; 68: 3645–3654.
- Peinado H, Olmeda D, Cano A.. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007; 7: 415–428.
- Batlle E, Sancho E, Francí C, et al. The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000; 2: 84–89.
- Cano A, Pérez-Moreno MA, Rodrigo I, et al. The transcription factor Snail controls epithelial–mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000; 2: 76–83.
- Vesuna F, van Diest P, Chen JH, et al. Twist is a transcriptional repressor of E-cadherin gene expression in breast cancer. Biochem Biophys Res Commun. 2008; 367: 235–241.
- Nieto MA. The Ins and Outs of the Epithelial to Mesenchymal Transition in Health and Disease. Annu Rev Cell Dev Biol. 2011; 27: 347–376.
- Bolós V, Peinado H, Pérez-Moreno MA, et al. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci. 2003; 116: 499–511.
- Sides MD, Klingsberg RC, Shan B, et al. The Epstein-Barr Virus Latent Membrane Protein 1 and Transforming Growth Factor–β1 Synergistically Induce Epithelial–Mesenchymal Transition in Lung Epithelial Cells. Am J Respir Cell Mol Biol. 2011; 44: 852–862.
- Zhong Q, Zhou B, Ann DK, et al. Role of Endoplasmic Reticulum Stress in Epithelial–Mesenchymal Transition of Alveolar Epithelial Cells: Effects of Misfolded Surfactant Protein. Am J Respir Cell Mol Biol. 2011; 45: 498–509.
- Tanjore H, Cheng DS, Degryse AL, et al. Alveolar Epithelial Cells Undergo Epithelial-to-Mesenchymal Transition in Response to Endoplasmic Reticulum Stress. J Biol Chem. 2011; 286: 30972–30980.
- Yao L, Conforti F, Hill C, et al. Paracrine signalling during ZEB1-mediated epithelial–mesenchymal transition augments local myofibroblast differentiation in lung fibrosis. Cell Death Differ. 2018; 1–15. doi:10.1038/s41418-018-0175-7
- Madtes DK, Busby HK, Strandjord TP, et al. Expression of transforming growth factor-alpha and epidermal growth factor receptor is increased following bleomycin-induced lung injury in rats. Am J Respir Cell Mol Biol. 1994; 11: 540–551.
- Liu JY, Morris GF, Lei WH, et al. Up-regulated expression of transforming growth factor-alpha in the bronchiolar-alveolar duct regions of asbestos-exposed rats. Am J Pathol. 1996; 149: 205–17.
- Waheed S, D'Angio CT, Wagner CL, et al. Transforming Growth Factor Alpha (Tgfα) Is Increased During Hyperoxia And Fibrosis. Exp Lung Res. 2002; 28: 361–372.
- Stella GM, Inghilleri S, Pignochino Y, et al. Activation of Oncogenic Pathways in Idiopathic Pulmonary Fibrosis. Transl Oncol. 2014; 7: 650–655.
- Tzouvelekis A, Ntolios P, Karameris A, et al. Increased expression of epidermal growth factor receptor (EGF-R) in patients with different forms of lung fibrosis. Biomed Res Int. 2013; 2013: 654354.
- Korfhagen TR, Swantz RJ, Wert SE, et al. Respiratory epithelial cell expression of human transforming growth factor-alpha induces lung fibrosis in transgenic mice. J Clin Invest. 1994; 93: 1691–1699.
- Hardie WD, Bruno MD, Huelsman KM, et al. Postnatal lung function and morphology in transgenic mice expressing transforming growth factor-alpha. Am J Pathol. 1997; 151: 1075–83.
- Madtes DK, Elston AL, Hackman RC, et al. Transforming Growth Factor- α Deficiency Reduces Pulmonary Fibrosis in Transgenic Mice. Am J Respir Cell Mol Biol. 1999; 20: 924–934.
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003; 3: 11–22.
- Wang Y, Ngo VN, Marani M, et al. Critical role for transcriptional repressor Snail2 in transformation by oncogenic RAS in colorectal carcinoma cells. Oncogene. 2010; 29: 4658–4670.
- Chilosi M, Caliò A, Rossi A, et al. Epithelial to mesenchymal transition-related proteins ZEB1, β-catenin and β-tubulin-III in idiopathic pulmonary fibrosis. Mod Pathol. 2017; 30: 26–38.
- Park JS, Park HJ, Park YS, et al. Clinical significance of mTOR, ZEB1, ROCK1 expression in lung tissues of pulmonary fibrosis patients. BMC Pulm Med. 2014; 14: 168.
- Rawlins EL, Hogan BLM, Emoto H, et al. Epithelial stem cells of the lung: privileged few or opportunities for many? Development. 2006; 133: 2455–65.
- King TE Jr, Schwarz MI, Brown K, et al. Idiopathic pulmonary fibrosis: Relationship between histopathologic features and mortality. Am J Respir Crit Care Med. 2001; 164: 1025–1032.
- Lomas NJ, Watts KL, Akram KM, et al. Idiopathic pulmonary fibrosis: immunohistochemical analysis provides fresh insights into lung tissue remodelling with implications for novel prognostic markers. Int J Clin Exp Pathol. 2012; 5: 58–71.
- Willis BC, Liebler JM, Luby-Phelps K, et al. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. 2005; 166: 1321–1332.
- Harada T, Nabeshima K, Hamasaki M, et al. Epithelial–mesenchymal transition in human lungs with usual interstitial pneumonia: Quantitative immunohistochemistry. Pathol Int. 2010; 60: 14–21.
- Marmai C, Sutherland RE, Kim KK, et al. Alveolar epithelial cells express mesenchymal proteins in patients with idiopathic pulmonary fibrosis. AJP Lung Cell Mol Physiol. 2011; 301: L71–L78.
- Kim KK, Wei Y, Szekeres C, et al. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Invest. 2009; 119: 213–224.
- Kim KK, Kugler MC, Wolters PJ, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A. 2006; 103: 13180–5.
- Tanjore H, Xu XC, Polosukhin VV, et al. Contribution of Epithelial-derived Fibroblasts to Bleomycin-induced Lung Fibrosis. Am J Respir Crit Care Med. 2009; 180: 657–665.
- Degryse AL, Tanjore H, Xu XC, et al. TGFβ signaling in lung epithelium regulates bleomycin-induced alveolar injury and fibroblast recruitment. Am J Physiol Lung Cell Mol Physiol. 2011; 300: L887-97.
- Degryse AL, Tanjore H, Xu XC, et al et al. Repetitive intratracheal bleomycin models several features of idiopathic pulmonary fibrosis. Am J Physiol Cell Mol Physiol. 2010; 299: L442–L452.
- DeMaio L, Buckley ST, Krishnaveni MS, et al. Ligand-independent transforming growth factor-β type I receptor signalling mediates type I collagen-induced epithelial-mesenchymal transition. J Pathol. 2012; 226: 633–644.
- Humphreys BD, Lin SL, Kobayashi A, et al. Fate Tracing Reveals the Pericyte and Not Epithelial Origin of Myofibroblasts in Kidney Fibrosis. Am J Pathol. 2010; 176: 85–97.
- Rock JR, Barkauskas CE, Cronce MJ, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci. 2011; 108: E1475–E1483.
- Grande MT, Sánchez-Laorden B, López-Blau C, et al. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med. 2015; 21: 989–997.
- Lovisa S, LeBleu VS, Tampe B, et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med. 2015; 21: 998–1009.
- Rowe RG, Lin Y, Shimizu-Hirota R, et al. Hepatocyte-Derived Snail1 Propagates Liver Fibrosis Progression. Mol Cell Biol. 2011; 31: 2392–2403.
- Hu K, Wu C, Mars WM, et al. Tissue-type plasminogen activator promotes murine myofibroblast activation through LDL receptor–related protein 1–mediated integrin signaling. J Clin Invest. 2007; 117: 3821–32.
- Xu Y, Mizuno T, Sridharan A, et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight. 2017; 1: e90558.
- Sakata J, Utsumi F, Suzuki S, et al. Inhibition of ZEB1 leads to inversion of metastatic characteristics and restoration of paclitaxel sensitivity of chronic chemoresistant ovarian carcinoma cells. Oncotarget. 2017; 8: 99482–99494.
- Kothari AN, Mi Z, Zapf M, et al. Novel clinical therapeutics targeting the epithelial to mesenchymal transition. Clin Transl Med. 2014; 3: 35.