
Combination therapy in Phe508del CFTR: how many will be enough?
Ana Margarida Matos1,2 and Paulo Matos1,2*
1Department of Human Genetics, National Health Institute ‘Dr. Ricardo Jorge’, Av. Padre Cruz, 1649-016 Lisboa; Portugal
2University of Lisboa; Faculty of Sciences, BioFIG - Centre for Biodiversity, Functional and Integrative Genomics, Campo Grande-C8, 1749-016 Lisboa; Portugal
Cystic fibrosis (CF) is a complex inherited disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene1. Around 2000 disease causing mutations are known for this gene, which encodes a Chloride (Cl−) channel expressed at the plasma membrane (PM) of epithelial cells1,2. Clinically the disease affects mostly the respiratory tract, where obstruction of the airways by thick mucus leads to: bacterial infections, extensive lung damage, and eventually, respiratory failure. Other affected systems include the gastrointestinal and reproductive tracts and endocrine system. Thus, the severity of symptoms can differ widely within individuals depending on their mutations, environment and biometrical characteristics. These variables modify the clinical course of the disease and each patient response to therapy3,4.
Current therapies are typically focused on treating CF multi-organ symptoms, as opposed to targeting the underling defect specific to each mutation5,6. This has been the goal of several CF drug discovery programs, either by companies or academic labs6,7, including our own8,9. This review aims to highlight the most recent therapies that target the molecular defect in CFTR, particularly the most common CFTR mutation worldwide, the deletion of phenylalanine 508 (Phe508del)10,11.
It is estimated that approximately 85% of all CF patients have at least 1 copy of the Phe508del mutant10,11. This mutation is characterized by defective protein processing, resulting in considerable endoplasmic reticulum (ER) retention and premature degradation, preventing the mutant protein from trafficking to the cell surface. The Phe508del-CFTR molecules that reach the cell surface present partial channel function and a highly decreased PM half-life, due to accelerated endocytosis and fast turnover5,12. Hence, applying strategies to correct Phe508del-CFTR multiple functional defects is complex, as more than one type of CFTR-modulator drug has to be used13,14.
At present, there are several CFTR-modulator drug combinations and exciting new next-generation CFTR modulators under study for the clinical treatment of CF patients with the Phe508del mutation7,15 (see Fig. 1). The most important will be addressed below.
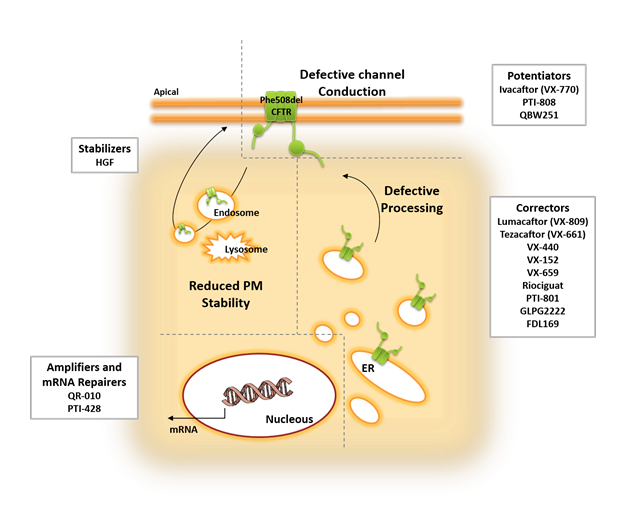
Figure 1: Overview of the several molecules and strategies currently under study to rescue the multiple defects of Phe508del CFTR.
Combination therapy
Lumacaftor plus Ivacaftor (Orkambi®)
Ivacaftor, also known as VX-770, is labeled as a CFTR potentiator, as it increases the time that activated CFTR channels remain open at the cell surface16. It was first introduced for the treatment of the Gly551Asp CFTR mutation, where it increased predicted mean forced expiratory volume in one second (FEV117, which in patients decreases from ~90% in infancy to ~50% by the age of 40 years-old18) by 10%, and was associated with less risk of pulmonary exacerbations and weight gain16. Since any Phe508del-CFTR protein reaching the cell surface also presents reduced channel function, the efficacy of Ivacaftor was assessed in subjects homozygous for the Phe508del mutation. In these phase II studies, the difference in the change of FEV1 % and other spirometric parameters did not reach statistical significance, thus, indicating that a CFTR potentiator alone is insufficient for the treatment of patients who are homozygous for the Phe508del genotype19,20.
Lumacaftor, also known as VX-809, is an established CFTR corrector drug that has been extensively characterized. Although the correction mechanism of Lumacaftor is not fully understood, there is evidence that it promotes the proper folding of Phe508del-CFTR during its biogenesis and processing in the ER, allowing it to exit the ER and traffic to the cell surface21–24. Improvement of CFTR function to clinically meaningful levels was proposed to require a combination of Lumacaftor, to deliver CFTR channels to the PM, and Ivacaftor, to increase the proportion of time those channels are open25. Based on this knowledge, Lumacaftor advanced into clinical trials in patients homozygous and heterozygous for the Phe508del mutation, with the aim of evaluating its safety and efficacy alone and in combination with Ivacaftor. As assessed by a phase II study, administration of Lumacaftor alone did not provide a significant therapeutic benefit, as predicted FEVM1 % was similar between the studied groups26. Subsequently, the combination of Ivacaftor and Lumacaftor was explored in a series of clinical studies. In a phase II study, combination of Lumacaftor and Ivacaftor (in the higher doses administrated) significantly improved FEV1 by a mean of 6% for patients homozygous for Phe508del CFTR, decreased sweat chloride concentration by a mean of 8.9 to 10.3 mmol/L, and decreased pulmonary exacerbations in the treatment groups27. Phe508del CFTR heterozygous patients did not have a significant improvement in FEV1 or any other parameters27. Data from two phase III trials in patients homozygous for Phe508del showed that there were significant improvements in FEV1, ranged from a mean of 2.6 to 4.0%, and that the rate of pulmonary exacerbations was 30 to 39% lower, since hospitalization or the use of intravenous antibiotics was reduced in the treatment groups28. While significant, these results fell below initial expectations and experimental evidence emerged to, at least partially, explain the limited improvements observed in patients. It was shown that chronic administration of Ivacaftor, as well as most other potentiators, results in a dose-dependent reversal of Lumacaftor- and Tezacaftor (another investigational corrector, see below)-mediated CFTR correction in Phe508del homozygous primary airway cell cultures20,29. This was due to protein destabilization and increase turnover rate, resulting in its reduced functional expression at the cell surface. A posterior study confirmed that exposure to high concentrations (>1 μM) of ivacaftor did inhibit lumacaftor’s rescue of Phe508del-CFTR but reported that chronic exposure to low (?100 nM), clinically relevant concentrations of the potentiator did not30. Thus, since combination therapy presumes an improvement of beneficial effects over the use of each drug alone, the inhibitory effect of ivacaftor on lumacaftor efficacy requires further evaluation, perhaps in post-treatment patient samples. In addition, it was also observed that P. aeruginosa reduces Phe508del-CFTR function in cells treated either with Lumacaftor alone or the with the Ivafactor/Lumacaftor combination31,32. Since 85% of adult CF patients are colonized with P. aeruginosa, this data suggests that infection with these bacteria may also partially account for a reduction in the therapeutic efficacy of these drugs in Phe508del-homozigous patients.
Nevertheless, despite its modest efficacy, the combination therapy clinical trial data supported an apparent benefit for patients. Accordingly, Ivacaftor plus Lumacaftor (commercial name: Orkambi®33) was approved for the clinical treatment of CF patients homozygous for the Phe508del mutation by the Food and Drug Administration (FDA) and European Medicines Agency (EMA)34. This drug combination has now been used in patients since 2015, and several consequent studies of its long-term usage indicate that it does benefit CF patients, although several cases of off-target side-effects have been reported, including dyspnea (14%), diarrhea (11%), and nausea (10%) as well as serious adverse hepatobiliary reactions occurring in at least 0.5% of patients35–37.
Tezacaftor plus Ivacaftor
Tezacaftor, also known as VX-661, is an investigational CFTR corrector, structurally similar to Lumacaftor, which also improves Phe508del CFTR folding and traffic to the cell surface38. Tezacaftor was introduced in clinical trials as an alternative to Lumacaftor, with the advantage that it is not an inducer of CYP3A4 enzymes and, therefore, does not interfere with other medications that are frequently used in CF, particularly Ivacaftor33,39. The safety and efficacy of Tezacaftor monotherapy, and Tezacaftor plus Ivacaftor combination therapy was evaluated in a phase II trial in patients homozygous for Phe508del or heterozygous for Phe508del and a second Gly551Asp CFTR mutation39. Administration of the combined therapy resulted in a significantly decrease in sweat chloride around 6.04 mmol/L and predicted FEV1 of 3.75% for Phe508del homozygous subjects, and 7.02 mmol/L in sweat chloride and 4.6% predicted FEV1 for heterozygous39. These results supported continued clinical studies of this drug combination, since the improvements in lung function are comparable to those observed in patients treated with lumacaftor plus Ivacaftor28. Results from phase III studies were similar, with values of 4.0% for predicted FEV1 in homozygous patients, with a 35% reduction on the rate of pulmonary exacerbation in the treatment group and 6.8% for predicted FEV1 in heterozygous patients40,41. In both phase II and III studies the treatment had less respiratory adverse events40,41 when compared with previous reports from Lumacaftor trials35–37, reveling itself to be an appealing alternative to the approved therapy.
Triple combinations
Last year Vertex announced the first results for the triple combination studies with Tezacaftor/Ivacaftor plus VX-440, VX-152, or VX-659, three investigational drugs that are next-generation correctors of the defective Phe508del-CFTR protein42–45. Data from the Phase II studies showed values of mean predicted FEV1 of 9.7% and 12.0% for the triple combination regimens with VX-152 or VX-440 respectively, in patients heterozygous for Phe508del and one minimal function mutation. In the same category of CF patients, initial data from a phase I study for the triple combination regimen of VX-659 showed an improvement in predicted FEV1 of 9.6%. Initial data with VX-152 or VX-440 in patients homozygous for Phe508del, who were already receiving Tezacaftor and Ivacaftor, also showed an improvement in mean predicted FEV1 of 7.3% and 9.5%. Furthermore, it was reported that all triple combinations were generally well tolerated and the majority of adverse events were mild to moderate42.
Plasma membrane stabilizers
Although preliminary results for the triple combination therapy in patients with the Phe508del mutation look promising, there is still an unmet target for truly effective new therapies. Given the complexity of protein defects presented in Phe508del CFTR, part of the incomplete effectiveness of the described combined therapies may derive from an inability to retain sufficient CFTR levels at the apical surface of epithelial cells. Therefore, there is a real need for molecular strategies achieving the PM retention of corrected rescued Phe508del CFTR. Our group has investigated the peripheral protein quality control (PPQC) checkpoint in lung epithelial cells in Phe508del CFTR exposed to VX-809. We found that the conformation of the scaffold protein NHERF1 (Na(+)/H(+) exchange regulatory factor 1) determined whether or not the PPQC recognized rescued Phe508del CFTR at the PM9. Moreover, we showed that activation of the cytoskeletal regulator Rac1 promoted an interaction between the actin-binding adaptor protein ezrin and NHERF1 in a way that triggered exposure of the second PDZ domain of NHERF1, which interacted with rescued Phe508del CFTR. Because binding of Phe508del CFTR to the second PDZ of NHERF1 precluded the recruitment of ubiquitin ligase CHIP, the co-exposure of airway cells to the Rac1 activator HGF (hepatocyte growth factor) nearly tripled the functional rescue of Phe508del CFTR by VX-809, retaining the rescued channels at the PM8. Since HGF signaling is determinant for lung tissue repair after acute lung injury46, these findings open new areas of investigation worth pursuing in the development of small-molecule drugs for CF treatment.
mRNA repairers
ProQR Therapeutics is initiating clinical trials for a novel mRNA-based strategy for correction of Phe508del CFTR mutation7 after obtaining extremely promising in vitro and in vivo preclinical data47. Their innovative compound, QR-010, is an investigational 33mer chemically modified, antisense oligoribonucleotide (AON) complementary to wt-CFTR mRNA, that was designed to repair Phe508del CFTR transcripts. QR-010 mechanism of action is not yet fully characterized but it was postulated to involve RNAse H-mediated degradation of the mutant transcript, followed by RNA repair48. Treatment with inhaled QR-010 brought near complete restoration of chloride transport across the nasal mucosa of Phe508del homozygous mice. Moreover, two doses of QR-010 were able to improve CFTR-mediated saliva secretion up to 80% of the wild-type levels47.
Proteostasis regulators
Proteostasis regulators are a novel class of CFTR correctors that act indirectly on CFTR by modulating components of the channel’s interactome. One of the most promising was Cavosonstat7, developed by Nivalis Therapeutics, Inc. Cavosonstat is a S-nitrosoglutathione reductase (GSNOR) inhibitor that increases GSNO and NO levels, which are lower in CF tissues49. This was postulated to promote a chaperone-dependent increase in CFTR abundance, stability and function. Unfortunately, in a February 2017 press-release, the company announced that the drug did not meet primary endpoints in a Phase II trial50. Nonetheless, it highlighted NO signaling as a new research venue for CFTR modulators.
Another NO-related proteostasis regulator is Riociguat, the active ingredient of Adempas, an oral drug marketed by Bayer for the treatment of pulmonary hypertension. Riociguat increases the sensitivity of soluble guanylate cyclase (sGC) to NO, increasing cyclic guanosine monophosphate (cGMP) production. In addition to decreasing blood pressure, some studies report that the sGC-NO-cGMP pathway may regulate CFTR channel conductivity51. A Phase 2 study is now underway in CF patients homozygous for the Phe508del mutation7.
Amplifiers
Amplifiers are compounds designed to increase CFTR expression and thus increase ER protein load. In that sense, to support Phe508del CFTR correction, they must be used in combination therapy. The starting point for the development of this CF drug class was the finding that the small-molecule HDAC7 inhibitor, SAHA1, significantly amplified Phe508del CFTR quantity and surface expression in human bronchial epithelial cells, possibly by interfering with CFTR’s proteostasis network52. However, the clinical applicability of HDAC inhibitors in CF treatment remains controversial. In a recent study using air-liquid interface cultures of differentiated nasal epithelia cells from CF patients, SAHA1 failed to increase CFTR transcript levels but rather inhibited mucine expression and goblet epithelial cell differentiation53. Nonetheless, those initial findings opened a new field in CF modulator drugs’ research. Proteostasis Therapeutics, Inc., for instance, has developed a specific CFTR amplifier, PTI-428, that increases the CFTR mRNA pool, feasibly by improving mRNA stability and/or events surrounding CFTR translation. A phase II study evaluating the efficacy and safety of PTI-428 in CF patients receiving Orkambi®33 demonstrated a mean absolute improvement in predicted FEV1 of 5.2% in the tested group, with no significant adverse effects54.
Additional small-molecule compounds
Proteostasis Therapeutics, Inc., developed two additional investigational compounds selectively targeting Phe508del CFTR: PTI-801, a new-generation CFTR corrector, and PTI-808, a CFTR potentiator7. The company just finished the first Phase I and II studies for these compounds, which showed promising results, and therefore, is now preparing new clinical studies and cohorts for the triple combination of PTI-428, PTI-801, and PTI-808, called PTI-NC-73354.
Galapagos/AbbVie has also developed four novel correctors currently under clinical trials7. Correctors GLPG2222 and GLPG2851 (C1) are additive to GLPG2737 and GLPG3221 (C2) and may be combined in therapy. GLPG2222 shares structural similarities with Lumacaftor and Tezacaftor, but was reported to produce a more efficient rescue of Phe508del CFTR in vitro55.
FDL169, a drug being developed by Flatley Discovery Lab, has been shown to increase Phe508del CFTR cell-surface abundance with similar potency and efficacy as VX-809. However, FDL169 activity is not additive to VX-809, suggesting a similar mode of action56. Notwithstanding, FDL169 exhibited a higher free fraction in human serum and improved distribution in mice lungs57, which makes it a promising alternative to Lumacaftor.
QBW251, a compound developed by Novartis Pharmaceuticals, is currently in phase II clinical trials7. It is described as a CFTR potentiator, similar to Ivacaftor. Preclinical data indicated that, when combined with Lumacaftor, QBW251 was more efficacious than Ivacaftor in sustaining Phe508del CFTR membrane expression and function58. In an initial study in CF patients homozygous and compound-heterozygous for the Phe508del mutation, QBW251 proved to be safe and well tolerated. It promoted a significant decrease in sweat chloride and improved lung function in heterozygous patients, but, as Ivacaftor, showed no efficacy in Phe508del homozygous individuals58. Nevertheless, these initial results suggest QBW251 may constitute an attractive alternative to Ivacaftor in combination therapies.
Notably, several other compounds were reported to potentiate CFTR function, including the natural food components genistein, an isoflavonoid found in high concentrations in soy59, and curcumin, a major constituent of turmeric60. Curcumin is able to activate CFTR channels in both adenosine triphosphate (ATP) dependent and independent ways60–62, whereas genistein acts through ATP-dependent CFTR gating63. Patch clamp studies showed additive effects of curcumin and genistein on the gating of Gly551Asp CFTR channels64 and, importantly, a recent study showed that genistein and curcumin also enhanced forskolin-induced swelling of ivacaftor/lumacaftor treated intestinal organoids derived from biopsies of Phe508del homozygous patients65. Curcumin PKA-dependent, but ATP-independent, potentiation results from its binding to the stimulatory ICL1/ICL4-R interface to stabilize the channel open state, while PKA/ATP-dependent potentiation results from removal of inhibitory Fe3+ at the ICL3-R interface, which promotes dimerization of NBD1 and NBD262,66–68. These data suggest that ivacaftor, genistein, and curcumin, in double or triple combinations, can synergize to restore CFTR-mediated fluid secretion in primary CF cells, thus supporting a possible benefit if multiple potentiators are used for treatment of CF. Still on this note, the very hydrophobic nature and low stability in water of compounds, such as curcumin, is a major drawback in their clinical application. In a very recent work, Gonçalves et al.69 showed that the neutral amphiphilic triblock copolymer MeOx6-THF19-MeOx6 (TBCP2) can solubilize curcumin and facilitate its penetration in Phe508del CFTR human airway epithelial cells, enhancing curcumin potentiation of CFTR mediate Cl- selective currents in these cells. These data suggests that TBCP2 may constitute a helpful tool for the delivery of this and other highly insoluble therapeutic drugs to CF patients.
Gene Therapy
Gene therapy is a controversial subject that became a possibility for CF since the cloning of the CFTR gene1. In the case of CF, direct administration of the agents to the lungs via aerosols is an attractive option for gene therapy as pulmonary epithelium has a relative easy accessibility and reduces the risks associated with systemic delivery. Non-viral vectors seem to be the safest and most efficient gene transfer agents for this kind of therapy and, in the most promising trials, the plasmid DNA pGM169 (carrying the CFTR cDNA) combined with the cationic liposome GL67A, was the chosen formula for lung delivery70,71. Monthly application of the pGM169/GL67A complex in CF patients was evaluated in a phase IIb trial, where a 3,7% increase in predicted FEV1 and stabilization of lung function was observed in the treatment group71. Despite the modest results, the trial offered proof of concept that non-viral gene therapy can benefit the lung function of CF patients and creates the opportunity for follow-up studies, namely combining gene therapy with the co-administration of CFTR modulator drugs.
Concluding Remarks
In the field of CF personalized medicine it is imperative to always consider the multidimensional nature of the disease. Even for CF patients caring the Phe508del mutation, a universal treatment is not likely to become a reality, given the complexity of phenotypes observed among the several possible compound-genotypes and even among homozygous patients. The contribution of lung tissue integrity, modifier genes, environmental factors, etc., has to be accounted, while tuning drug combination therapies to each individual patient. This is even more relevant if one considers therapies to fit the plethora of CFTR mutations displaying distinct functional defects. Nonetheless, the data presented in this review support a direct association between targeting the various defects of Phe508del-CFTR with combinations of multiple pharmaceutical agents and a consistent gain in lung function, body weight, and reduced disease exacerbations in patients. Thus, these recent achievements with small-molecule therapies, coupled to the growing amount of novel compounds and innovative strategies, give the scientific, clinical and patient communities new hope of finding highly efficient therapeutically solutions for CF.
Conflict of interest statement
The authors declare no conflict of interest.
Funding information
The authors have been supported by grants IF2012 (from FTC, Portugal) and PGG-055-2014 (from Gilead Genése, Portugal) to PM and by BioISI center grant UID/MULTI/04046/2013 (from FCT/MCTES/PIDDAC, Portugal). AMM is recipient of FCT fellowship SFRH/BD/524906/2014.
Abbreviations:
AON :Oligoribonucleotide; ATP : Adenosine Triphosphate; CF : Cystic Fibrosis; CFTR : Cystic Fibrosis Transmembrane Conductance Regulator; cGMP : cyclic guanosine monophosphate; Chloride : Cl-; EMA : European Medicines Agency; ER : Endoplasmic Reticulum; FDA : Food and Drug Administration; FEV1 : Forced Expiratory Volume in One Second; GSNOR : S-nitrosoglutathione reductase; HGF : Hepatocyte Growth Factor; ICL : Intracellular Loop; NHERF1 : Na(+)/H(+) Exchange Regulatory Factor 1; Phe508del : Deletion of phenylalanine 508; PM : Plasma Membrane; PPQC : Peripheral Protein Quality Control; R : Regulatory; sGC : Soluble Guanylate Cyclase; TBCP2 : MeOx6-THF19-MeOx6.
References
- Riordan JR, Rommens JM, Kerem B, et al. Identification of the Cystic Fibrosis Gene?: Cloning and Characterization of Complementary DNA. Science (80- ). 1989; 245(4922): 1066–73.
- Sosnay PR, Siklosi KR, Van Goor F, et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat Genet [Internet]. 2013 Aug 25 [cited 2017 Dec 28]; 45(10): 1160–7. Available from: http://www.nature.com/doifinder/10.1038/ng.2745
- About Cystic Fibrosis | CF Foundation [Internet]. [cited 2017 Dec 28]. Available from: https://www.cff.org/What-is-CF/About-Cystic-Fibrosis/
- Collins FS. Cystic fibrosis: molecular biology and therapeutic implications. Science [Internet]. 1992 May 8 [cited 2017 Dec 26]; 256(5058): 774–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/1375392
- Amaral MD. Novel personalized therapies for cystic fibrosis: treating the basic defect in all patients. J Intern Med [Internet]. 2015 Feb [cited 2017 Dec 28]; 277(2): 155–66. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25266997
- Ashlock MA, Olson ER. Therapeutics Development for Cystic Fibrosis: A Successful Model for a Multisystem Genetic Disease. Annu Rev Med [Internet]. Annual Reviews; 2011 Feb 18 [cited 2017 Dec 29]; 62(1): 107–25. Available from: http://www.annualreviews.org/doi/10.1146/annurev-med-061509-131034
- Drug Development Pipeline | CFF Clinical Trials Tool [Internet]. [cited 2018 Jan 2]. Available from: https://www.cff.org/trials/pipeline
- Sousa M, Moraes BJ, Mendes AI, et al. HGF Stimulation of Rac1 Signaling Enhances Pharmacological Correction of the Most Prevalent Cystic Fibrosis Mutant F508del- CFTR. 2012;
- Loureiro CA, Matos AM, Dias-Alves A, et al. A molecular switch in the scaffold NHERF1 enables misfolded CFTR to evade the peripheral quality control checkpoint. Sci Signal [Internet]. 2015 May 19 [cited 2015 May 20]; 8(377): ra48-ra48. Available from: http://stke.sciencemag.org/content/8/377/ra48.full
- Welcome to CFTR2 | CFTR2 [Internet]. [cited 2017 Dec 28]. Available from: https://www.cftr2.org/
- De Boeck K, Zolin A, Cuppens H, et al. The relative frequency of CFTR mutation classes in European patients with cystic fibrosis. J Cyst Fibros [Internet]. Elsevier; 2014 Jul 1 [cited 2017 Dec 28]; 13(4): 403–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24440181
- De Boeck K, Amaral MD. Progress in therapies for cystic fibrosis. Lancet Respir Med [Internet]. Elsevier; 2016 Aug 1 [cited 2017 Dec 28]; 4(8): 662–74. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27053340
- D Amaral M, M Farinha C. Rescuing Mutant CFTR: A Multi-task Approach to a Better Outcome in Treating Cystic Fibrosis. Curr Pharm Des [Internet]. 2013 Apr 1; 19(19): 3497–508. Available from: http://www.eurekaselect.com/openurl/content.php?genre=article&issn=1381-6128&volume=19&issue=19&spage=3497
- Farinha CM, Matos P. Repairing the basic defect in cystic fibrosis - one approach is not enough. FEBS J [Internet]. 2016 Jan 1 [cited 2018 Jan 8]; 283(2): 246–64. Available from: http://doi.wiley.com/10.1111/febs.13531
- Smith CA, Collins CT, Cyna AM, et al. Complementary and alternative therapies for pain management in labour. In: Smith CA, editor. Cochrane Database of Systematic Reviews [Internet]. Chichester, UK: John Wiley & Sons, Ltd; 2006 [cited 2017 Jul 13]. p. CD003521. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17054175
- Ramsey BW, Davies J, McElvaney NG, et al. A CFTR Potentiator in Patients with Cystic Fibrosis and the G551D Mutation. N Engl J Med [Internet]. Massachusetts Medical Society ; 2011 Nov 3 [cited 2017 Dec 12]; 365(18): 1663–72. Available from: http://www.nejm.org/doi/abs/10.1056/NEJMoa1105185
- Konstan MW, Wagener JS, Yegin A, et al. Design and powering of cystic fibrosis clinical trials using rate of FEV(1) decline as an efficacy endpoint. J Cyst Fibros [Internet]. Elsevier; 2010 Sep 1 [cited 2017 Dec 15]; 9(5): 332–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20646968
- Kerem E, Viviani L, Zolin A, et al. Factors associated with FEV1 decline in cystic fibrosis: analysis of the ECFS Patient Registry. Eur Respir J [Internet]. 2014 Jan 1 [cited 2018 Jan 18]; 43(1): 125–33. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23598952
- Flume Pa, Liou TG, Borowitz DS, et al. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest [Internet]. 2012 Sep [cited 2014 Oct 14]; 142(3): 718–24. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3435140&tool=pmcentrez&rendertype=abstract
- Cholon DM, Quinney NL, Fulcher ML, et al. Potentiator ivacaftor abrogates pharmacological correction of ΔF508 CFTR in cystic fibrosis. Sci Transl Med [Internet]. American Association for the Advancement of Science; 2014 Jul 23 [cited 2017 Dec 22]; 6(246): 246ra96. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25101886
- Van Goor F, Hadida S, Grootenhuis PDJ, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A [Internet]. 2011 Nov 15 [cited 2014 Sep 13]; 108(46): 18843–8. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3219147&tool=pmcentrez&rendertype=abstract
- Ren HY, Grove DE, De La Rosa O, et al. VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Mol Biol Cell [Internet]. 2013 Oct [cited 2015 May 8]; 24(19): 3016–24. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3784376&tool=pmcentrez&rendertype=abstract
- Farinha CM, King-Underwood J, Sousa M, et al. Revertants, low temperature, and correctors reveal the mechanism of F508del-CFTR rescue by VX-809 and suggest multiple agents for full correction. Chem Biol [Internet]. 2013 Jul 25 [cited 2014 Oct 21]; 20(7): 943–55. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23890012
- Okiyoneda T, Veit G, Dekkers JF, et al. Mechanism-based corrector combination restores ΔF508-CFTR folding and function. Nat Chem Biol [Internet]. Nature Publishing Group; 2013 May 12 [cited 2018 Jan 2]; 9(7): 444–54. Available from: http://www.nature.com/doifinder/10.1038/nchembio.1253
- Sloane PA, Rowe SM. Cystic fibrosis transmembrane conductance regulator protein repair as a therapeutic strategy in cystic fibrosis. Curr Opin Pulm Med [Internet]. 2010 Nov [cited 2014 Oct 20]; 16(6): 591–7. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3733473&tool=pmcentrez&rendertype=abstract
- Clancy JP, Rowe SM, Accurso FJ, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax [Internet]. BMJ Publishing Group Ltd; 2012 Jan 1 [cited 2017 Dec 22]; 67(1): 12–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21825083
- Boyle MP, Bell SC, Konstan MW, et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med [Internet]. Elsevier; 2014 Jul 1 [cited 2017 Dec 14]; 2(7): 527–38. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24973281
- Wainwright CE, Elborn JS, Ramsey BW, et al. Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med [Internet]. Massachusetts Medical Society; 2015 Jul 16 [cited 2017 Dec 12]; 373(3): 220–31. Available from: http://www.nejm.org/doi/10.1056/NEJMoa1409547
- Veit G, Avramescu RG, Perdomo D, et al. Some gating potentiators, including VX-770, diminish F508-CFTR functional expression. Sci Transl Med [Internet]. 2014; 6(246): 246ra97-246ra97. Available from: http://stm.sciencemag.org/content/6/246/246ra97.abstract
- Matthes E, Goepp J, Carlile GW, et al. Low free drug concentration prevents inhibition of F508del CFTR functional expression by the potentiator VX-770 (ivacaftor). Br J Pharmacol [Internet]. 2015 Oct 22 [cited 2015 Nov 16]; Available from: http://www.readcube.com/articles/10.1111%2Fbph.13365
- Stanton BA. Effects of Pseudomonas aeruginosa on CFTR chloride secretion and the host immune response. Am J Physiol - Cell Physiol [Internet]. 2017 Apr 1 [cited 2017 Dec 15]; 312(4): C357–66. Available from: http://ajpcell.physiology.org/lookup/doi/10.1152/ajpcell.00373.2016
- Stanton BA, Coutermarsh B, Barnaby R, et al. Pseudomonas aeruginosa Reduces VX-809 Stimulated F508del-CFTR Chloride Secretion by Airway Epithelial Cells. Ahmad S, editor. PLoS One [Internet]. Public Library of Science; 2015 May 27 [cited 2018 Jan 30]; 10(5): e0127742. Available from: http://dx.plos.org/10.1371/journal.pone.0127742
- ORKAMBI® (lumacaftor/ivacaftor) Tablets [Internet]. [cited 2018 Jan 2]. Available from: https://www.orkambi.com/
- Our Approved Medicines | Vertex Pharmaceuticals [Internet]. [cited 2017 Dec 29]. Available from: https://www.vrtx.com/pipeline-medicines/our-approved-medicines
- Hubert D, Chiron R, Camara B, et al. Real-life initiation of lumacaftor/ivacaftor combination in adults with cystic fibrosis homozygous for the Phe508del CFTR mutation and severe lung disease. J Cyst Fibros [Internet]. Elsevier; 2017 May 1 [cited 2017 Dec 15]; 16(3): 388–91. Available from: https://www.sciencedirect.com/science/article/pii/S1569199317300863
- Konstan MW, McKone EF, Moss RB, et al. Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): a phase 3, extension study. Lancet Respir Med [Internet]. Elsevier; 2017 Feb 1 [cited 2017 Dec 15]; 5(2): 107–18. Available from: https://www.sciencedirect.com/science/article/pii/S2213260016304271
- Jennings MT, Dezube R, Paranjape S, et al. An Observational Study of Outcomes and Tolerances in Patients with Cystic Fibrosis Initiated on Lumacaftor/Ivacaftor. Ann Am Thorac Soc [Internet]. 2017 Nov [cited 2018 Jan 4]; 14(11): 1662–6. Available from: http://www.atsjournals.org/doi/10.1513/AnnalsATS.201701-058OC
- Eckford PDW, Ramjeesingh M, Molinski S, et al. VX-809 and Related Corrector Compounds Exhibit Secondary Activity Stabilizing Active F508del-CFTR after Its Partial Rescue to the Cell Surface. Chem Biol [Internet]. 2014 [cited 2018 Jan 4]; 21: 666–78. Available from: http://dx.doi.org/10.1016/j.chembiol.2014.02.021
- Donaldson SH, Pilewski JM, Griese M, et al. Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am J Respir Crit Care Med [Internet]. 2017 Sep 20 [cited 2018 Jan 4];rccm.201704-0717OC. Available from: http://www.atsjournals.org/doi/10.1164/rccm.201704-0717OC
- Rowe SM, Daines C, Ringshausen FC, et al. Tezacaftor–Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N Engl J Med [Internet]. Massachusetts Medical Society; 2017 Nov 23 [cited 2017 Dec 12]; 377(21): 2024–35. Available from: http://www.nejm.org/doi/10.1056/NEJMoa1709847
- Taylor-Cousar JL, Munck A, McKone EF, et al. Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N Engl J Med [Internet]. Massachusetts Medical Society; 2017 Nov 23 [cited 2017 Dec 12]; 377(21): 2013–23. Available from: http://www.nejm.org/doi/10.1056/NEJMoa1709846
- Vertex Announces Positive Phase 1 & Phase 2 Data from Three Different Triple Combination Regimens in People with Cystic Fibrosis Who Have One F508del Mutation and One Minimal Function Mutation (F508del/Min) (NASDAQ:VRTX) [Internet]. [cited 2017 Dec 14]. Available from: http://investors.vrtx.com/releasedetail.cfm?releaseid=1033559
- A Study Evaluating the Safety and Efficacy of VX-440 Combination Therapy in Subjects With Cystic Fibrosis - Full Text View - ClinicalTrials.gov [Internet]. [cited 2017 Dec 12]. Available from: https://clinicaltrials.gov/ct2/show/NCT02951182
- A Study Evaluating the Safety of VX-152 Combination Therapy in Adults With Cystic Fibrosis - Full Text View - ClinicalTrials.gov [Internet]. [cited 2018 Dec 2]. Available from: https://clinicaltrials.gov/ct2/show/NCT02951195
- A Study to Evaluate Safety and Pharmacokinetics of VX-659 in Healthy Subjects and in Adults With Cystic Fibrosis - Full Text View - ClinicalTrials.gov [Internet]. [cited 2017 Dec 12]. Available from: https://clinicaltrials.gov/ct2/show/NCT03029455
- Zeng L, Yang X, Li H, et al. The cellular kinetics of lung alveolar epithelial cells and its relationship with lung tissue repair after acute lung injury. Respir Res [Internet]. 2016 Dec 7 [cited 2018 Jan 8]; 17(1): 164. Available from: http://respiratory-research.biomedcentral.com/articles/10.1186/s12931-016-0480-y
- Beumer W, Swildens J, Henig N, et al. WS01.2 QR-010, an RNA therapy, restores CFTR function using in vitro and in vivo models of ΔF508 CFTR. J Cyst Fibros [Internet]. Elsevier; 2015 Jun 1 [cited 2018 Jan 8]; 14: S1. Available from: https://www.sciencedirect.com/science/article/pii/S1569199315300023
- Zamecnik PC, Raychowdhury MK, Tabatadze DR, et al. Reversal of cystic fibrosis phenotype in a cultured 508 cystic fibrosis transmembrane conductance regulator cell line by oligonucleotide insertion. Proc Natl Acad Sci [Internet]. 2004 May 25 [cited 2018 Jan 18]; 101(21): 8150–5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15148387
- Solomon GM, Marshall SG, Ramsey BW, et al. Breakthrough therapies: Cystic fibrosis (CF) potentiators and correctors. Pediatr Pulmonol [Internet]. 2015 Oct [cited 2018 Jan 8]; 50(S40): S3–13. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26097168
- Nivalis Therapeutics Announces Results from Phase 2 Clinical Trial of Cavosonstat Added to Ivacaftor for Treatment of Cystic Fibrosis Nasdaq:NVLS [Internet]. [cited 2018 Jan 8]. Available from: https://globenewswire.com/news-release/2017/02/23/927232/0/en/Nivalis-Therapeutics-Announces-Results-from-Phase-2-Clinical-Trial-of-Cavosonstat-Added-to-Ivacaftor-for-Treatment-of-Cystic-Fibrosis.html
- Grasemann H, Gaston B, Fang K, et al. Decreased levels of nitrosothiols in the lower airways of patients with cystic fibrosis and normal pulmonary function. J Pediatr [Internet]. 1999 Dec [cited 2018 Jan 8]; 135(6): 770–2. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10586185
- Hutt DM, Herman D, Rodrigues APC, et al. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat Chem Biol [Internet]. 2010 Jan 6 [cited 2018 Jan 8]; 6(1): 25–33. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19966789
- Bergougnoux A, Petit A, Knabe L, et al. The HDAC inhibitor SAHA does not rescue CFTR membrane expression in Cystic Fibrosis. Int J Biochem Cell Biol [Internet]. 2017 Jul [cited 2018 Jan 18]; 88: 124–32. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28478266
- Proteostasis Therapeutics Announces Progression of PTI-428 and PTI-801 to Longer Duration Studies in CF Subjects | Proteostasis Therapeutics [Internet]. [cited 2018 Jan 8]. Available from: http://ir.proteostasis.com/news-releases/news-release-details/proteostasis-therapeutics-announces-progression-pti-428-and-pti
- Wang X, Liu B, Searle X, et al. Discovery of 4-[(2 R ,4 R )-4-({[1-(2,2-Difluoro-1,3-benzodioxol-5-yl)cyclopropyl]carbonyl}amino)-7-(difluoromethoxy)-3,4-dihydro-2 H -chromen-2-yl]benzoic Acid (ABBV/GLPG-2222), a Potent Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Corrector for the Treatment of Cystic Fibrosis. J Med Chem [Internet]. 2018 Jan 5 [cited 2018 Jan 9];acs.jmedchem.7b01339. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29251932
- Mijnders M, Kleizen B, Braakman I. Correcting CFTR folding defects by small-molecule correctors to cure cystic fibrosis. Curr Opin Pharmacol [Internet]. Elsevier; 2017 Jun 1 [cited 2018 Jan 8]; 34: 83–90. Available from: https://www.sciencedirect.com/science/article/pii/S1471489217301285
- Zawistoski M, Sui J, Ordonez C, et al. 32 Properties of a novel F508del-CFTR corrector FDL169. J Cyst Fibros [Internet]. Elsevier; 2016 Jun 1 [cited 2018 Jan 18]; 15: S59–60. Available from: http://linkinghub.elsevier.com/retrieve/pii/S1569199316302727
- QBW251 Is a Safe and Efficacious CFTR Potentiator for Patients with Cystic Fibrosis. Available from: http://www.atsjournals.org/doi/abs/10.1164/ajrccm-conference.2016.193.1_MeetingAbstracts.A7789
- French PJ, Bijman J, Bot AG, et al. Genistein activates CFTR Cl- channels via a tyrosine kinase- and protein phosphatase-independent mechanism. Am J Physiol Physiol [Internet]. 1997 Aug [cited 2018 Jan 18]; 273(2): C747–53. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9277373
- Wang W, Bernard K, Li G, et al. Curcumin Opens Cystic Fibrosis Transmembrane Conductance Regulator Channels by a Novel Mechanism That Requires neither ATP Binding nor Dimerization of the Nucleotide-binding Domains. J Biol Chem [Internet]. 2007 Feb 16 [cited 2018 Jan 18]; 282(7): 4533–44. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17178710
- Eckford PDW, Li C, Ramjeesingh M, et al. Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Potentiator VX-770 (Ivacaftor) Opens the Defective Channel Gate of Mutant CFTR in a Phosphorylation-dependent but ATP-independent Manner. J Biol Chem [Internet]. 2012 Oct 26 [cited 2018 Jan 18]; 287(44): 36639–49. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22942289
- Wang G. The Inhibition Mechanism of Non-phosphorylated Ser 768 in the Regulatory Domain of Cystic Fibrosis Transmembrane Conductance Regulator. J Biol Chem [Internet]. 2011 Jan 21 [cited 2018 Jan 30]; 286(3): 2171–82. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21059651
- Moran O, Galietta LJ V, Zegarra-Moran O. Binding site of activators of the cystic fibrosis transmembrane conductance regulator in the nucleotide binding domains. C Cell Mol Life Sci [Internet]. 2005 Feb [cited 2018 Jan 18]; 62(4): 446–60. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15719171
- Sohma Y, Yu YC, Hwang TC. Curcumin and genistein: the combined effects on disease-associated CFTR mutants and their clinical implications. Curr Pharm Des [Internet]. 2013 [cited 2018 Jan 18]; 19(19): 3521–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23331029
- Dekkers JF, Van Mourik P, Vonk AM, et al. Potentiator synergy in rectal organoids carrying S1251N, G551D, or F508del CFTR mutations. J Cyst Fibros [Internet]. 2016 Sep [cited 2018 Jan 18]; 15(5): 568–78. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27160424
- Wang G. Molecular Basis for Fe(III)-Independent Curcumin Potentiation of Cystic Fibrosis Transmembrane Conductance Regulator Activity. Biochemistry [Internet]. 2015 May 12 [cited 2018 Feb 5]; 54(18): 2828–40. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25867080
- Wang G, Linsley R, Norimatsu Y. External Zn 2+ binding to cysteine-substituted cystic fibrosis transmembrane conductance regulator constructs regulates channel gating and curcumin potentiation. FEBS J [Internet]. 2016 Jul 1 [cited 2018 Jan 31]; 283(13): 2458–75. Available from: http://doi.wiley.com/10.1111/febs.13752
- Wang G. Interplay between Inhibitory Ferric and Stimulatory Curcumin Regulates Phosphorylation-Dependent Human Cystic Fibrosis Transmembrane Conductance Regulator and ΔF508 Activity. Biochemistry [Internet]. 2015 Feb 24 [cited 2018 Feb 5]; 54(7): 1558–66. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25635662
- Gonçalves C, Gomez J-P, Même W, et al. Curcumin/poly(2-methyl-2-oxazoline-b-tetrahydrofuran-b-2-methyl-2-oxazoline) formulation: An improved penetration and biological effect of curcumin in F508del-CFTR cell lines. Eur J Pharm Biopharm [Internet]. 2017 Aug [cited 2018 Jan 18]; 117: 168–81. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28427956
- Davies G, Davies JC, Gill DR, et al. T4 Safety and expression of a single dose of lipid-mediated CFTR gene therapy to the upper and lower airways of patients with Cystic Fibrosis. Thorax [Internet]. BMJ Publishing Group Ltd; 2011 Dec 1 [cited 2018 Jan 17]; 66(Suppl 4): A2–A2. Available from: http://thorax.bmj.com/cgi/doi/10.1136/thoraxjnl-2011-201054a.4
- Alton EWFW, Armstrong DK, Ashby D, et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir Med [Internet]. Elsevier; 2015 Sep [cited 2018 Jan 17]; 3(9): 684–91. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26149841